RNA-Seqデータ解析の流れ 〜DEG解析やGO/パスウェイ解析〜
はじめに
次世代シーケンサーを用いてRNA-Seq解析を行うと、FASTQファイルと呼ばれる生データが得られます。 この生データから遺伝子発現の定量や発現が変動した遺伝子の抽出(DEG解析)、発現に変動のあった遺伝子の機能解析(GO解析、パスウェイ解析)等を行うには、様々なデータ処理を実施する必要があります。
本ページでは、ヒトやマウス、ラットのようにアノテーションが充実してる生物種について生データからRNA-Seqのデータ解析を行うための手順を解説します。
大まかなデータ解析の流れ
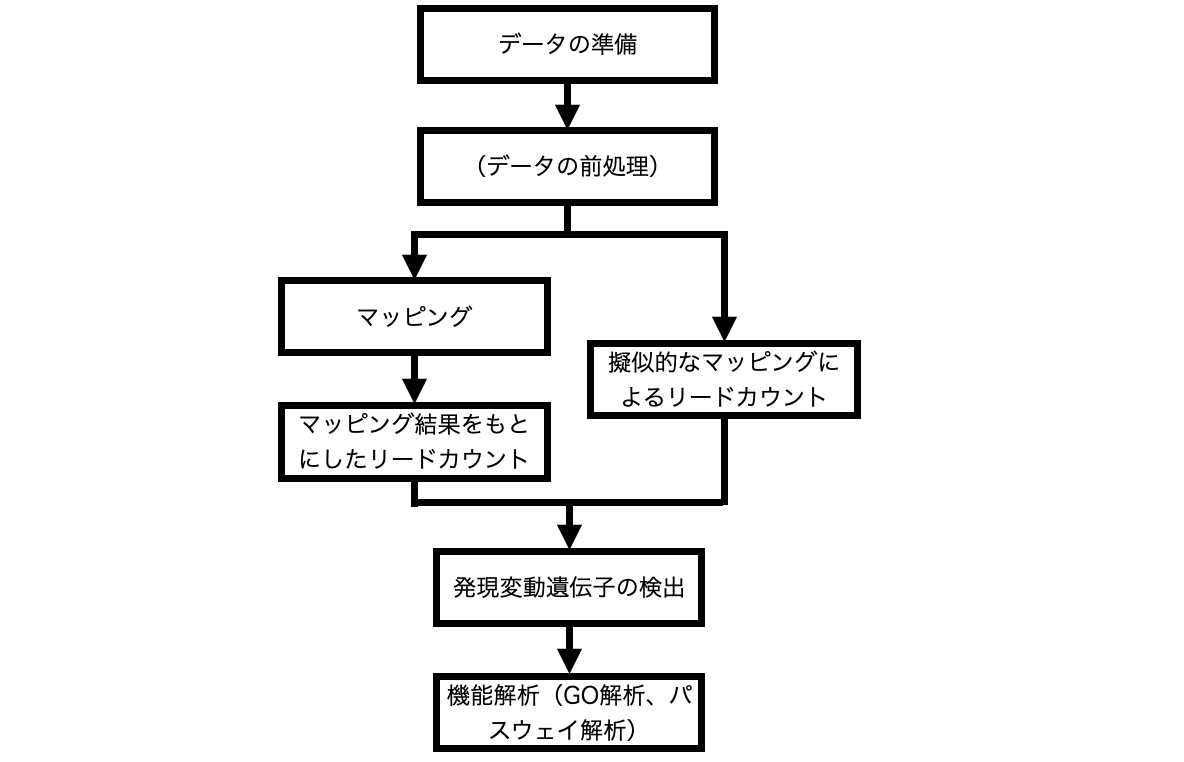
処理一覧
| 処理 | 概要 |
|---|---|
| データの準備 | 解析に用いるFASTQファイルを準備します。 |
| データの前処理 | FASTQファイルからアダプター配列をトリミングしたり、品質の悪いリードをフィルタリングしたりします。 |
| マッピング | リード配列を参照配列の塩基配列が一致する箇所に並べていきます。 |
| マッピング結果をもとにしたリードカウント | マッピング結果をもとに各遺伝子に何本のリードがマッピングされたかカウントします。 |
| 擬似的なマッピングによるリードカウント | FASTQファイルをもとに各遺伝子に何本のリードがマッピングされるかカウントします。 |
| 発現変動遺伝子の検出(DEG解析) | 異なる条件間で発現量に変化があった遺伝子を見つけます。 |
| 機能解析(GO解析、パスウェイ解析) | 見つかった遺伝子がどのような機能に関わっているか明らかにします。 |
データの準備
まずは、データ解析に用いるFASTQファイルを準備する必要があります。
FASTQファイルは次世代シーケンサーによるシーケンスで取得できます。 ご自身でシーケンスを実施したり、外注に出したりすることになるでしょう。 外注に出す場合には、一括見積ツールもございますので是非ご利用ください。
公共データベースからFASTQファイルをダウンロードすることもできます。 その場合にはfasterq-dumpというツールを使うのが良いでしょう。
fasterq-dumpのさらに詳しい説明はこちらをご覧ください。
関連ページ
データの前処理
次世代シーケンサーから出力される生データには、アダプター配列が含まれているリードや品質の悪いリードが存在しています。 そのため、FASTQファイルからアダプター配列をトリミングしたり、品質の悪いリードをフィルタリングしたりといったデータの前処理を実施することがあります。
FASTQファイルの前処理に使われるソフトウェアとしては、Trimmomatic、Cutadapt、FastQC、fastp等があります。
以下はfastpを用いて前処理を行った結果の例です。19,786,002リード、2,967,900,000塩基あったデータが19,722,456リード、2,941,557,000塩基になりました。
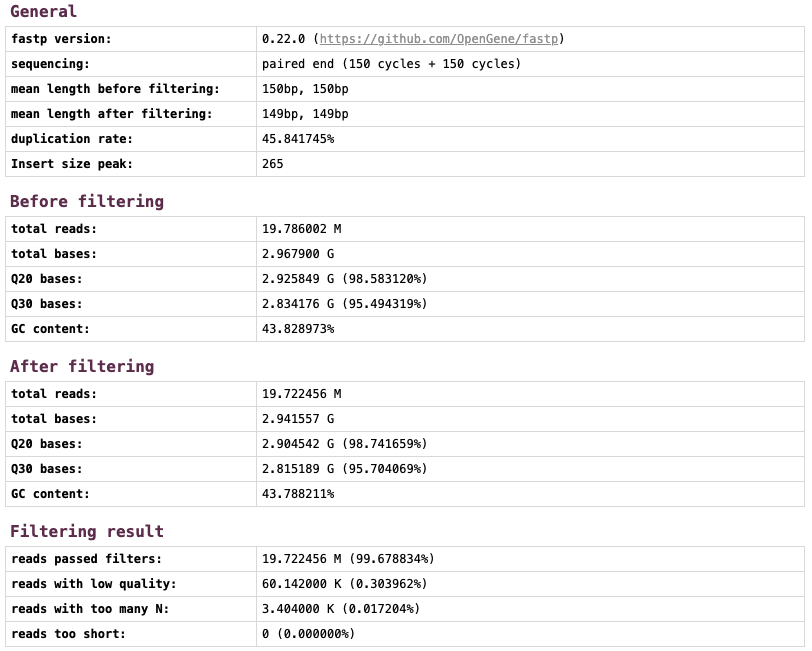
fastpのさらに詳しい説明はこちらをご覧ください。
関連ページ
マッピング
次に、次世代シーケンサーで得られたリード配列を参照配列にマッピングしていきます。 マッピングとはリード配列を参照配列の塩基配列が一致する箇所に並べていく解析を言います。
マッピング結果はBAMファイルと呼ばれる形式で出力されます。 つまり、マッピングとはFASTQファイルと参照配列をインプットとしてBAMファイルを得る解析ということができます。
マッピングによく使われるソフトウェアとして、HISAT2、STAR、Bowtie2といったものがあります。
マッピング結果を可視化すると以下のようなイメージとなります。
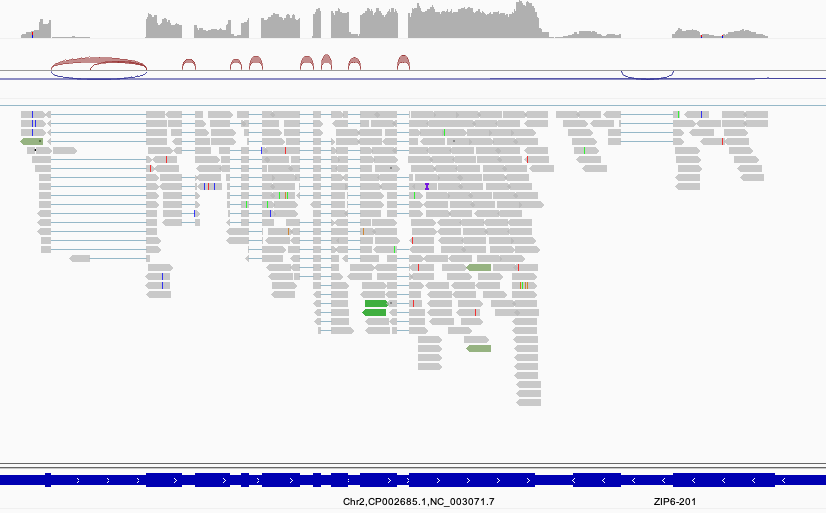
関連ページ
マッピング結果をもとにしたリードカウント
次に、マッピング結果から各遺伝子に何本のリードがマッピングされたかカウントしていきます。 この操作では、参照配列およびアノテーションファイル(GTFファイル, GFF3ファイル)、マッピング結果(BAMファイル)をもとにリードをカウントしていきます。
マッピング結果をもとにしたリードカウントにはfeatureCounts、htseq-count、RSEM、StringTieといったソフトウェアがよく使われます。 遺伝子レベルでのリードカウントであれば、featureCountsやhtseq-countで十分ですが、転写産物レベルでのリードカウントが必要な場合にはRSEMやStringTieを用います。
リードカウントを行うと以下のような結果が得られます。
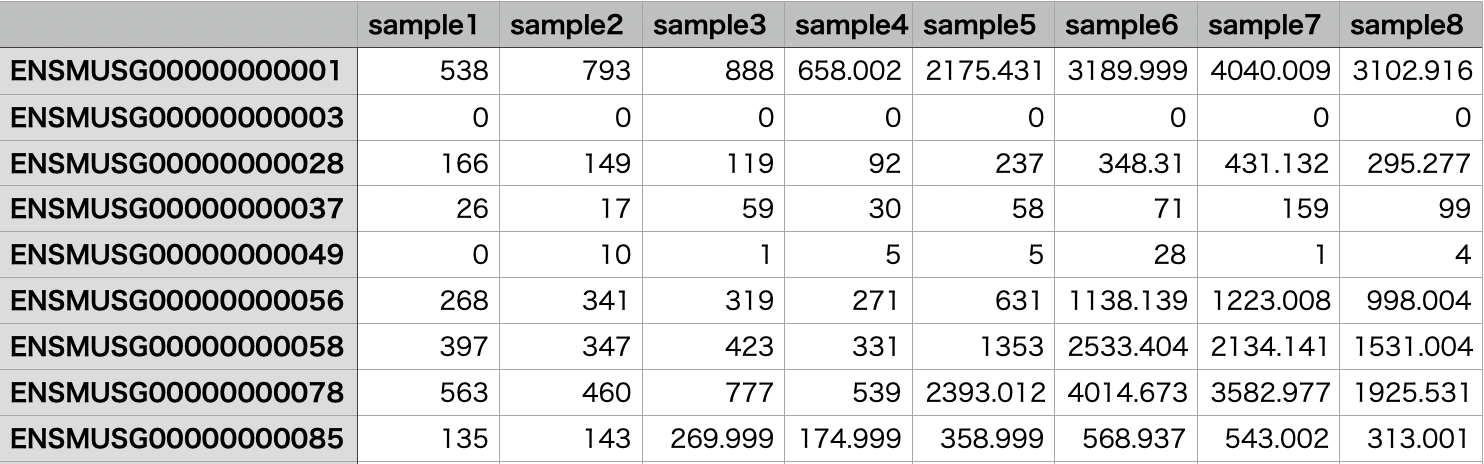
関連ページ
擬似的なマッピングによるリードカウント
マッピング結果をもとにしたリードカウントとは別の方法で、擬似的なマッピング処理によるリードカウントを行うこともあります。
この手法を用いる場合には事前にマッピング処理を行う必要はなく、 FASTQファイルと参照トランスクリプトーム配列をインプットとして非常に高速にリードカウント結果が得られます。
擬似的なマッピングによるリードカウントには、SalmonやKallistoといったソフトウェアがよく使われます。
擬似的なマッピングによるリードカウントでもマッピング結果をもとにしたリードカウントと同様の結果が得られます。
関連ページ
発現変動遺伝子の抽出(DEG解析)
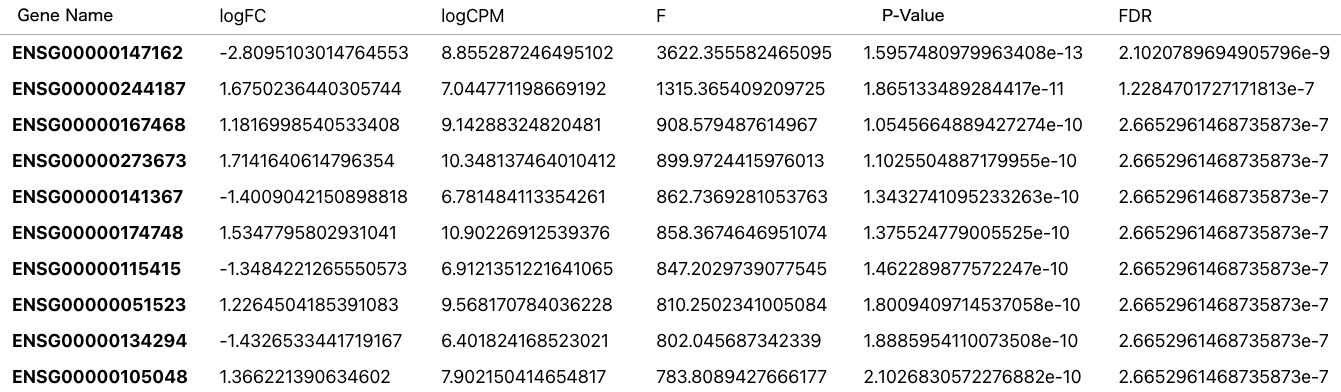
発現変動遺伝子(DEGs, Differentially Expressed Genes)とは、異なる条件やグループ間において発現量が大きく上昇または減少した遺伝子のことを言います。 リードカウントの結果をもとに発現変動遺伝子を抽出できます。 発現変動遺伝子の検出に使われるソフトウェアとして、Ballgown、edgeRやDESeq2といったものがあります。
発現変動遺伝子の抽出を行うと以下のような結果が得られます。
関連ページ
機能解析(GO解析、パスウェイ解析)
発現変動遺伝子の抽出(DEG解析)によって作成された遺伝子のリストに対して、どのような機能に多く関わっているかを調べる解析です。
遺伝子の機能を表現する方法として、Gene Ontologyやパスウェイが用いられることが多いです。
機能解析には、clusterProfilerやtopGO、GOseqといったソフトウェアがよく使われます。
以下はGO解析の結果の例です。

関連ページ
論文に必要な解析が簡単にできるRNA-Seqデータ解析ツール
RNA-Seqデータ解析ツールを利用すれば、外部委託や共同研究者への依頼は必要ありません。高スペックなコンピュータの準備やLinuxコマンドの操作も不要ですので、いますぐにご自身で解析できるようになります。
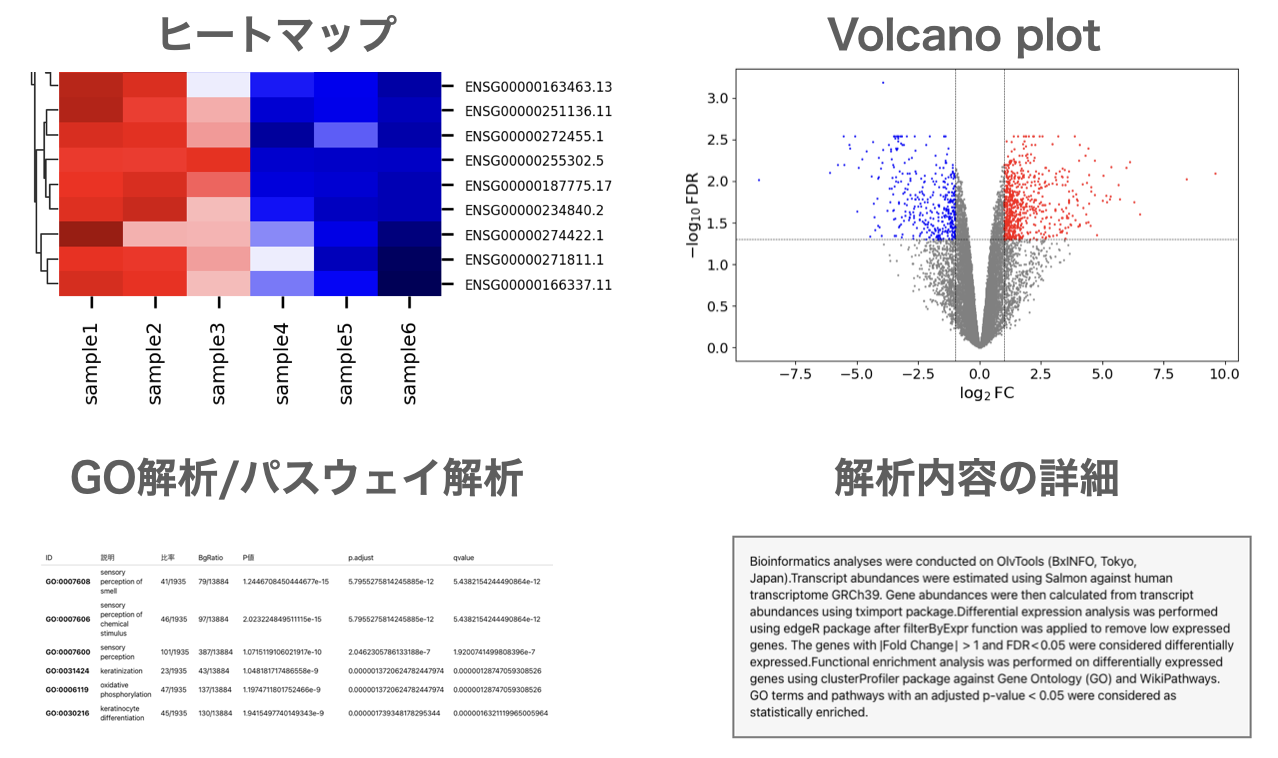
遺伝子発現量の定量、発現変動遺伝子抽出(DEG解析)、Volcano plot描画、MAプロット描画、ヒートマップ描画、GO解析、パスウェイ解析等 を簡単に実施できます。