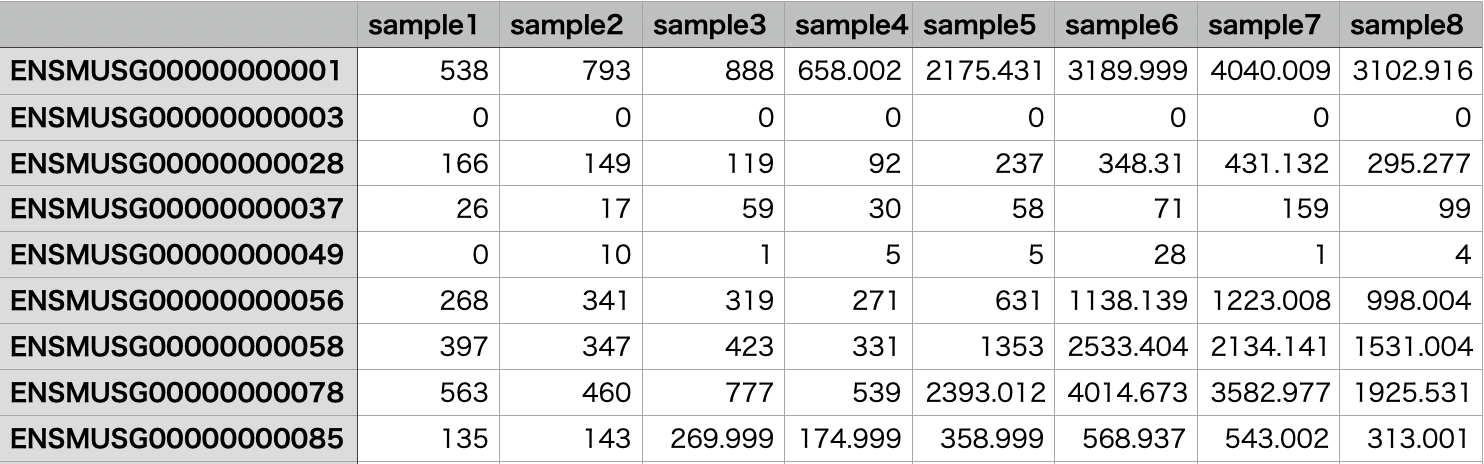
【edgeRの使い方】発現変動遺伝子の検出 edgeRとは 次世代シーケンサーを用いてRNA-Seq解析を行うとそれぞれの遺伝子の発現量が得られます。 複数のサンプルのRNA-Seq解析を行った際に、グループ間の遺伝子の発現量比較を行うことで発現が変動した遺伝子を見つけたいということがよくあります。
この発現変動遺伝子の検出に使われるソフトウェアがedgeRです。 本ページではedgeRの使い方やインストール方法を解説します。
以下の操作が難しいと感じる場合には、WEB上で簡単に発現変動遺伝子を検出できるツール も公開しておりますので、是非ご利用ください。
edgeRのインストール まずはじめに R がない場合には、R をインストールします。(Homebrewを使ったインストール例です。)
$ brew install r
Rを起動して、以下を実行して BiocManager および edgeR をインストールします。
> if (!requireNamespace("BiocManager", quietly=TRUE))
> install.packages("BiocManager")
> BiocManager::install("edgeR")
以下を実行して、エラーが表示されなければインストール成功です。
> library(edgeR)
データの準備 edgeRの使い方 > counts <- read.csv("counts.csv", sep=",", row.names=1)
> group <- factor(c("A", "A", "A", "A", "B", "B", "B", "B"))
> y <- DGEList(counts=counts, group=group)
カウントデータを読み込んで、グループの情報と合わせてDGEListオブジェクトを作成しました。 今回は、sample1~4とsample5~8の2群比較を行いたいので、AとBの2群に分けました。
> nrow(y)
[1] 35627
> keep <- filterByExpr(y)
> y <- y[keep, , keep.lib.sizes=FALSE]
> nrow(y)
[1] 14698
filterByExprを用いて発現の少ない遺伝子をフィルタリングしました。 この例では、遺伝子数が35,627から14,698に減少しました。
> y <- calcNormFactors(y)
> y$samples
group lib.size norm.factors
sample1 A 20190676 0.5252614
sample2 A 17815578 0.7264956
sample3 A 16858297 0.8385346
sample4 A 17080450 0.5972758
sample5 B 18305317 1.4576576
sample6 B 23123425 1.5620988
sample7 B 23260262 1.6452239
sample8 B 19145522 1.3967109
calcNormFactorsを用いてサンプル間のバイアスを補正するためのTMM正規化 を実施しました。
最後に、quasi-likelihood F-testsを実施します。
> design <- model.matrix(~group)
> y <- estimateDisp(y, design)
> fit <- glmQLFit(y, design)
> qlf <- glmQLFTest(fit, coef=2)
> topTags(qlf)
Coefficient: groupB
logFC logCPM F PValue FDR
ENSMUSG00000038738 -3.934239 4.352090 191.17683 4.406800e-08 0.0006477115
ENSMUSG00000021250 2.543736 6.859874 111.07055 6.409002e-07 0.0027772690
ENSMUSG00000032487 3.859926 1.891284 103.08209 9.187893e-07 0.0027772690
ENSMUSG00000070495 -3.377550 3.217322 99.99038 1.063542e-06 0.0027772690
ENSMUSG00000064356 -3.325373 11.863375 96.88620 1.237031e-06 0.0027772690
ENSMUSG00000033453 -1.469414 5.458883 95.42623 1.330180e-06 0.0027772690
ENSMUSG00000100862 -15.490014 7.031386 169.49446 1.993728e-06 0.0027772690
ENSMUSG00000096887 -3.332279 8.621508 85.88952 2.194494e-06 0.0027772690
ENSMUSG00000054942 -1.453327 5.837918 83.01079 2.577827e-06 0.0027772690
ENSMUSG00000004842 -5.072918 2.248245 82.51494 2.651651e-06 0.0027772690
発現変動遺伝子が抽出できました。以下で、FDR < 0.05の行を抽出できます。
> result <- as.data.frame(topTags(qlf, n=nrow(y)))
> result[result$FDR<0.05,]
logFC logCPM F PValue FDR
ENSMUSG00000038738 -3.9342393 4.35208965 191.17683 4.406800e-08 0.0006477115
ENSMUSG00000021250 2.5437356 6.85987389 111.07055 6.409002e-07 0.0027772690
ENSMUSG00000032487 3.8599263 1.89128388 103.08209 9.187893e-07 0.0027772690
ENSMUSG00000070495 -3.3775501 3.21732235 99.99038 1.063542e-06 0.0027772690
ENSMUSG00000064356 -3.3253735 11.86337535 96.88620 1.237031e-06 0.0027772690
ENSMUSG00000033453 -1.4694139 5.45888339 95.42623 1.330180e-06 0.0027772690
ENSMUSG00000100862 -15.4900144 7.03138596 169.49446 1.993728e-06 0.0027772690
ENSMUSG00000096887 -3.3322793 8.62150780 85.88952 2.194494e-06 0.0027772690
ENSMUSG00000054942 -1.4533272 5.83791761 83.01079 2.577827e-06 0.0027772690
ENSMUSG00000004842 -5.0729179 2.24824466 82.51494 2.651651e-06 0.0027772690
...
FDR < 0.05で抽出すれば、抽出された遺伝子の中で実際には変動していない遺伝子(偽陽性)の割合は5%ととなります。
TMM正規化とは TMM正規化とはRNA-Seq解析における遺伝子発現量の補正方法の1つで、edgeRに実装されている手法です。
RNA-Seqで測定できるのは、絶対的な発現量ではなく相対的な発現量です。 そのため、少ない遺伝子がたくさん発現している場合に、相対的に他の遺伝子の発現量が減少したように見えてしまいます。 そこでTMM正規化では、サンプル間の発現量の違いを最小化するように補正を行います。 この方法を用いれば、サンプル間で大部分の遺伝子の発現が変動していないデータであれば妥当な補正を行えることになります。
なお、TMM正規化ではサンプル間で共通の要素については補正を行いません。 例えば、遺伝子長はリードカウントに相関すると言われており、遺伝子長が長いほどリードカウントは多くなりますが、TMM正規化では補正していません。 edgeRでは発現変動遺伝子の抽出にフォーカスしており、遺伝子間の補正は必要ありませんので、TMM正規化で十分です。
一方で、FPKM/RPKM やTPM といった正規化手法では遺伝子間の発現量を比較することも考慮して遺伝子長に対する補正も行っています。
論文に必要な解析が簡単にできるRNA-Seqデータ解析ツール RNA-Seqデータ解析ツール を利用すれば、外部委託や共同研究者への依頼は必要ありません。高スペックなコンピュータの準備やLinuxコマンドの操作も不要ですので、いますぐにご自身で解析できるようになります。
遺伝子発現量の定量、発現変動遺伝子抽出(DEG解析)、Volcano plot描画、MAプロット描画、ヒートマップ描画、GO解析、パスウェイ解析等 を簡単に実施できます。
論文に必要な解析が簡単にできるRNA-Seqデータ解析ツールへ