【TopHat2の使い方】RNA-Seq解析におけるマッピング はじめに RNA-Seq解析によって得られたシーケンスデータを用いて遺伝子発現量を定量していく際に、一般的にマッピング処理が行われます。 マッピングとはリード配列 (FASTQファイル )を参照配列の一致する箇所に並べていく処理のことを言います。 RNA-Seqにおけるマッピングによく使われるソフトウェアとして、HISAT2 、STAR 、Bowtie2 といったものがあります。 本ページではTopHat2の使い方を説明します。
なお、TopHat2は古いソフトウェアですので、通常はHISAT2やSTARを使うことをおすすめします。
RNA-Seqのデータ解析の一連の流れはこちら をご覧ください。
インストール TopHat2を使うにはBowtie2のindexが必要になります。 Condaを利用してTopHat2をインストールすれば、自動的にBowtie2もインストールされます。
$ conda install -c bioconda tophat
Bowtie2のヘルプを表示してみます。
$ bowtie2 -h
以下のような内容が表示されれば成功です。
bowtie2 -h
Bowtie 2 version 2.4.1 by Ben Langmead (langmea@cs.jhu.edu, www.cs.jhu.edu/~langmea)
Usage:
bowtie2 [options]* -x <bt2-idx> {-1 <m1> -2 <m2> | -U <r> | --interleaved <i> | -b <bam>} [-S <sam>]
<bt2-idx> Index filename prefix (minus trailing .X.bt2).
NOTE: Bowtie 1 and Bowtie 2 indexes are not compatible.
<m1> Files with #1 mates, paired with files in <m2>.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<m2> Files with #2 mates, paired with files in <m1>.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<r> Files with unpaired reads.
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<i> Files with interleaved paired-end FASTQ/FASTA reads
Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).
<bam> Files are unaligned BAM sorted by read name.
<sam> File for SAM output (default: stdout)
<m1>, <m2>, <r> can be comma-separated lists (no whitespace) and can be
specified many times. E.g. '-U file1.fq,file2.fq -U file3.fq'.
...
TopHat2のヘルプを表示してみます。
$ tophat
以下のような内容が表示されれば成功です。
tophat
tophat:
TopHat maps short sequences from spliced transcripts to whole genomes.
Usage:
tophat [options] <bowtie_index> <reads1[,reads2,...]> [reads1[,reads2,...]] [quals1,[quals2,...]] [quals1[,quals2,...]]
Options:
-v/--version
-o/--output-dir <string> [ default: ./tophat_out ]
--bowtie1 [ default: bowtie2 ]
-N/--read-mismatches <int> [ default: 2 ]
--read-gap-length <int> [ default: 2 ]
--read-edit-dist <int> [ default: 2 ]
--read-realign-edit-dist <int> [ default: "read-edit-dist" + 1 ]
-a/--min-anchor <int> [ default: 8 ]
-m/--splice-mismatches <0-2> [ default: 0 ]
-i/--min-intron-length <int> [ default: 50 ]
-I/--max-intron-length <int> [ default: 500000 ]
-g/--max-multihits <int> [ default: 20 ]
--suppress-hits
-x/--transcriptome-max-hits <int> [ default: 60 ]
-M/--prefilter-multihits ( for -G/--GTF option, enable
an initial bowtie search
against the genome )
--max-insertion-length <int> [ default: 3 ]
--max-deletion-length <int> [ default: 3 ]
--solexa-quals
--solexa1.3-quals (same as phred64-quals)
--phred64-quals (same as solexa1.3-quals)
-Q/--quals
--integer-quals
-C/--color (Solid - color space)
--color-out
--library-type <string> (fr-unstranded, fr-firststrand,
fr-secondstrand)
-p/--num-threads <int> [ default: 1 ]
-R/--resume <out_dir> ( try to resume execution )
-G/--GTF <filename> (GTF/GFF with known transcripts)
--transcriptome-index <bwtidx> (transcriptome bowtie index)
-T/--transcriptome-only (map only to the transcriptome)
-j/--raw-juncs <filename>
--insertions <filename>
--deletions <filename>
-r/--mate-inner-dist <int> [ default: 50 ]
--mate-std-dev <int> [ default: 20 ]
--no-novel-juncs
--no-novel-indels
--no-gtf-juncs
--no-coverage-search
--coverage-search
--microexon-search
--keep-tmp
--tmp-dir <dirname> [ default: <output_dir>/tmp ]
-z/--zpacker <program> [ default: gzip ]
-X/--unmapped-fifo [use mkfifo to compress more temporary
files for color space reads]
...
index作成(build) まず以下のコマンドで参照配列のindexを作成していきます。こちらではBowtie2を使います。
$ bowtie2-build -f genome.fa genome
genome.faはマッピングしたい参照配列のFASTAファイル です。gzipで圧縮されたままでも問題ありません。
この操作によりgenome.1.bt2〜genome10.4.bt2とgenome.rev.1.bt2、genome.rev.2.bt2の6つのファイルが作成されます。 indexファイルは文字列を高速に検索するために必要なファイルで、Bowtie2に限らずほぼすべてのマッピングソフトウェアにおいて事前作成が必要です。
マッピング 次に、リード配列を参照配列にマッピングしていきます。こちらではTopHat2を使います。
$ tophat -o output genome reads1.fastq.gz reads2.fastq.gz
この操作により output ディレクトリ内に「accepted_hits.bam」という名前のBAMファイル が出力されました。
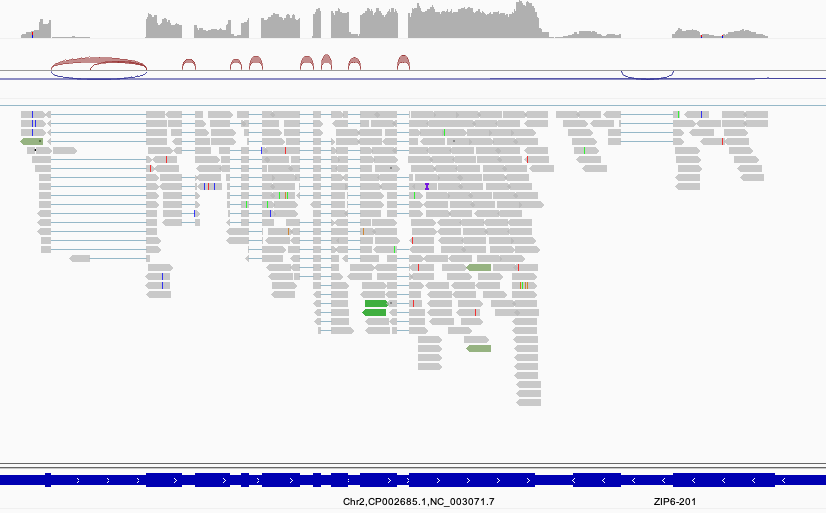
結果をIGV等のゲノムブラウザで可視化すると以下のようにマッピングされた様子が確認できます。
論文に必要な解析が簡単にできるRNA-Seqデータ解析ツール RNA-Seqデータ解析ツール を利用すれば、外部委託や共同研究者への依頼は必要ありません。高スペックなコンピュータの準備やLinuxコマンドの操作も不要ですので、いますぐにご自身で解析できるようになります。
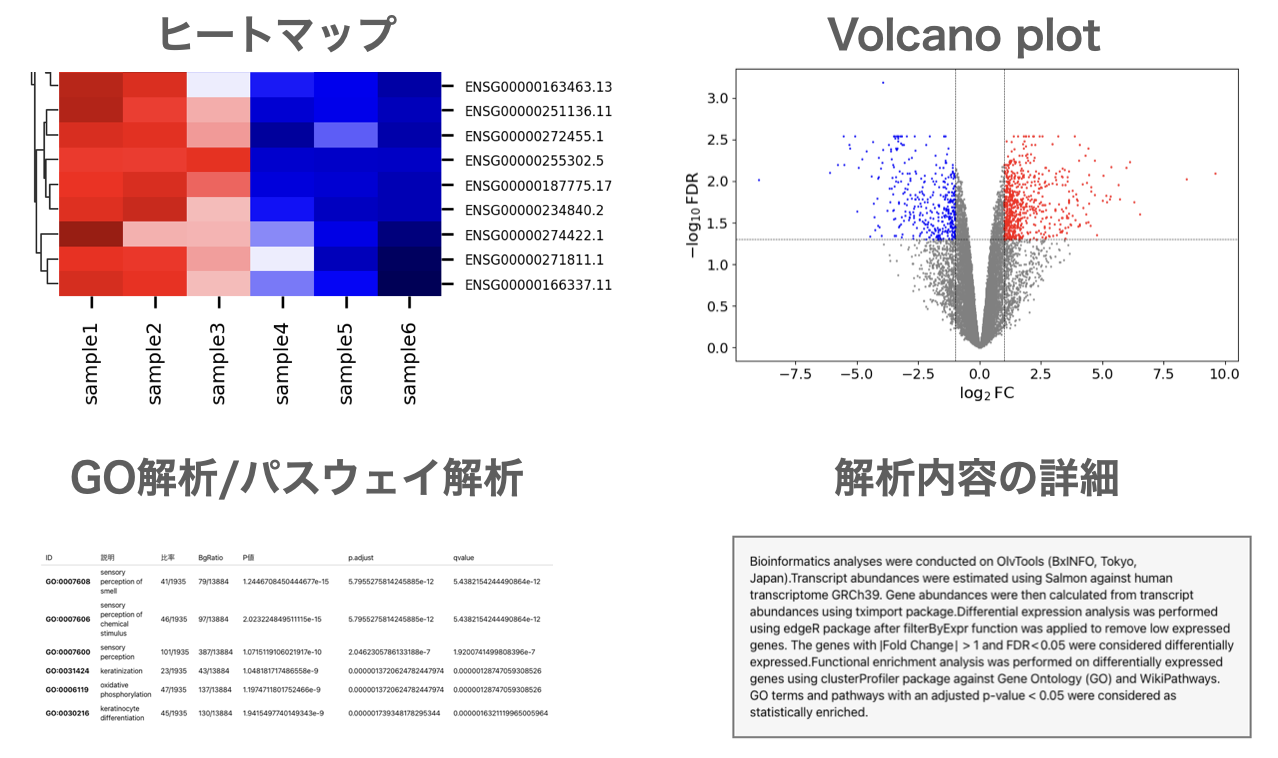
遺伝子発現量の定量、発現変動遺伝子抽出(DEG解析)、Volcano plot描画、MAプロット描画、ヒートマップ描画、GO解析、パスウェイ解析等 を簡単に実施できます。
論文に必要な解析が簡単にできるRNA-Seqデータ解析ツールへ