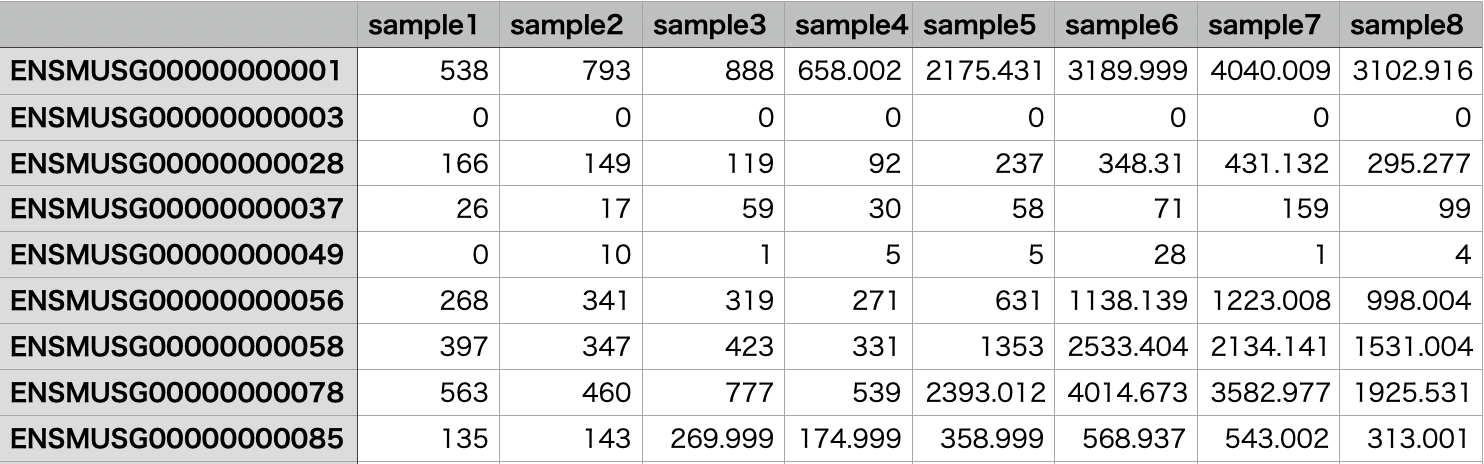
> res <- results(dds)
> res
log2 fold change (MLE): condition B vs A
Wald test p-value: condition B vs A
DataFrame with 62696 rows and 8 columns
baseMean log2FoldChange lfcSE stat pvalue padj
<numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000290825.1 0.0000 NA NA NA NA NA
ENSG00000223972.6 0.0000 NA NA NA NA NA
ENSG00000227232.5 11.4438 -0.619619 0.758925 -0.816443 0.414247 NA
ENSG00000278267.1 1.6048 -0.826152 1.974423 -0.418427 0.675635 NA
ENSG00000243485.5 0.0000 NA NA NA NA NA
... ... ... ... ... ... ...
ENSG00000198695.2 0 NA NA NA NA NA
ENSG00000210194.1 0 NA NA NA NA NA
ENSG00000198727.2 0 NA NA NA NA NA
ENSG00000210195.2 0 NA NA NA NA NA
ENSG00000210196.2 0 NA NA NA NA NA